
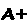
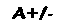
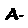

As hemoglobinas mutantes introduziram o conceito de patologia molecular, que se refere a doenças causadas pela alteração de uma única proteína. As mutações na molécula de hemoglobina ocasionam, freqüentemente, a substituição de um único aminoácido. O aminoácido modificado nas hemoglobinas mutantes pode ter localizações diversas, trazendo conseqüências variáveis.
Quando está localizado na superfície da molécula, sua alteração geralmente é inócua. A grande exceção a esta regra é a hemoglobina S (HbS), que causa a anemia falciforme. A frequência do gene de HbS, ao contrário dos genes de outras hemoglobinas mutadas, é alta, chegando a 40% em algumas regiões da África.
Como a sobrevida dos homozigotos para HbS é baixa, somente uma forte pressão seletiva poderia explicar a alta incidência do gene defectivo, isto é, o heterozigoto deveria ter alguma vantagem em relação ao homozigoto normal. De fato, os portadores do gene mutado são resistentes a uma forma letal de malária.
A incidência desta doença e a freqüência do gene para HbS são altamente correlacionadas. A anemia falciforme demonstra claramente que mutações deletérias podem constituir ferramentas da evolução, determinando uma maior probabilidade de sobrevivência de seus portadores.
Mutações em aminoácidos situados no interior da molécula geralmente determinam a síntese de hemoglobinas não-funcionais. A perda da função normal deve-se a diferentes conseqüências dessas mutações: estrutura instável, afinidade por oxigênio alterada ou oxidação do ferro (Fe2+) do grupo heme a Fe3+ .
Hemoglobinas instáveis, com estrutura espacial distorcida, podem resultar de ruptura de elementos da estrutura terciária, como acontece quando há substituição de um aminoácido por prolina, que interrompe a α-hélice. Em outros casos, a associação do grupo heme com a cadeia polipeptídica torna-se mais lábil, por causa da troca de um aminoácido com cadeia lateral apolar por outro com grupo R polar, o que altera o caráter hidrofóbico da cavidade onde se aloja o heme.
Alterações nas interfaces α1β2 e α2β1 entre os dímeros causam afinidade anormal por oxigênio. A afinidade é maior quando a mutação ocasiona rompimento de ligações que estabilizam a forma desoxigenada que, assim, mais facilmente se converte em oxiemoglobina; ao contrário, mutações que diminuem a estabilidade da forma oxigenada reduzem a afinidade por oxigênio.
A meta-hemoglobina (HbM) contém o ferro do grupo heme no estado férrico (Fe3+), que não se liga ao oxigênio. Esta alteração pode resultar da exposição a reagentes oxidantes ou de mutações. A formação espontânea de meta-hemoglobina e de metamioglobina explica a cor marrom característica de sangue desidratado e de carnes deterioradas.
A meta-hemoglobina (HbM) contém o ferro do grupo heme no estado férrico (Fe3+), que não se liga ao oxigênio. Esta alteração pode resultar da exposição a reagentes oxidantes ou de mutações. A formação espontânea de meta-hemoglobina e de metamioglobina explica a cor marrom característica de sangue desidratado e de carnes deterioradas.
Pacientes portadores de HbM são, geralmente, cianóticos: a pele e as mucosas exibem forte cor azulada, devido à concentração anormalmente elevada de desoxi-Hb na circulação arterial (a cor azulada das veias superficiais é uma conseqüência normal do seu alto conteúdo de desoxi-Hb). Esse tipo de anomalia só foi observada na forma heterozigota, certamente porque a homozigose seria fatal.
O segundo grupo de lesões genéticas é caracterizado pela síntese não-estequiométrica das subunidades da hemoglobina - são as talassemias, que têm alta incidência na região do Mar Mediterrâneo (thalassa, em grego, significa mar).
Nas α-talassemias, geralmente causadas por deleção gênica, a produção das cadeias α é defectiva, enquanto nas β-talassemias, resultantes de vários tipos de mutações, faltam as cadeias β.
Os homozigotos apresentam anemia severa e essa condição é denominada talassemia maior; os heterozigotos são assintomáticos (talassemia menor) e, como acontece na anemia falciforme, apresentam uma certa proteção contra a malária.
A hemoglobina pode sofrer, ainda, alterações químicas transitórias, resultantes de processos endógenos, como é o caso de sua glicosilação, ou da ação de drogas e poluentes ambientais.